
Im letzten Kapitel haben wir die berühmte Grundgleichung von Daniel Bernoulli für die statistische Interpretation der Bewegung von Gasmolekülen und letztlich der Temperatur hergeleitet:
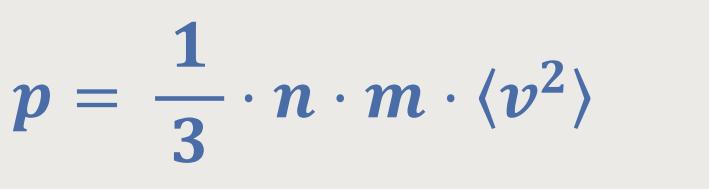
p ist der Druck auf die Wand des Behälters
n = N /V ist hier die die Molekülanzahldichte
m ist die Masse eines Moleküls
⟨ v²⟩
ist das mittlere Geschwindigkeitsquadrat der Moleküle
Mit wenigen weiteren Schritten kann aus dieser Gleichung abgeleitet werden, was physikalisch unter der Temperatur zu verstehen ist. Bis jetzt wurde die Temperatur nur rein empirisch ermittelt. Hierzu wurde zwischen den jeweiligen Schmelzpunkten entsprechend interpoliert. Aber das ist keine wirkliche Definition für eine physikalische Größe.
Wir erinnern uns, die Zustandsgleichung für ideale Gase lautete:
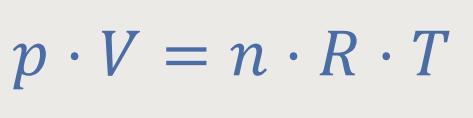
Und die Molekülanzahldichte war definiert als:
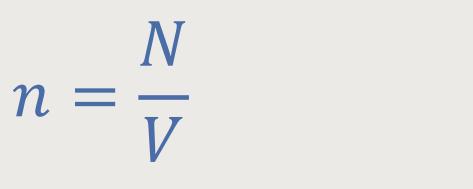
Wenn man jetzt die Bernoulli-Gleichung zunächst einmal mit dem Volumen V erweitert und gleichzeitig die Molekülanzahldichte berücksichtigt ergibt sich:
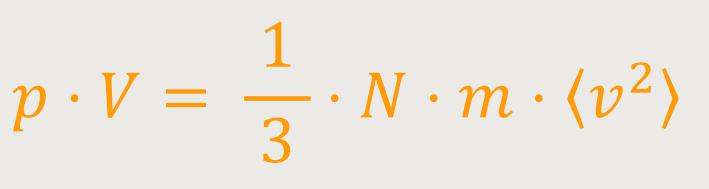
Außerdem wissen wir, dass die kinetische Energie eines Moleküls
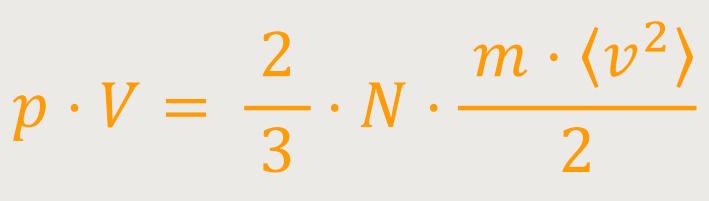
Auf diese Weise erhalten wir als Faktor den Mittelwert der kinetischen Energie von einem
Molekül. Natürlich kann ein solches Molekül auch Rotationen und innere
Schwingungen vollziehen. Dadurch wird dem Molekül weitere kinetische Energie
zugeführt. Und zwar kinetische Energie der Rotation und kinetische Energie der Schwingungen.
Wir hatten jedoch vorausgesetzt, dass es sich nur um punktförmige Teilchen
handeln soll, die sich vorerst nur translatorisch fortbewegen. In diesem Fall steht
der Ausdruck
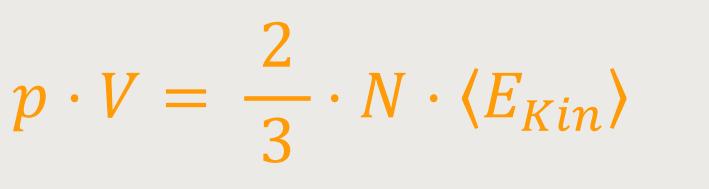
Daraus erkennt man schon, dass die mittlere kinetische Energie eines Moleküls proportional zur Temperatur des Körpers ist:

Und jetzt macht man einen entsprechenden Ansatz, um beides miteinander in Relation zu bringen:
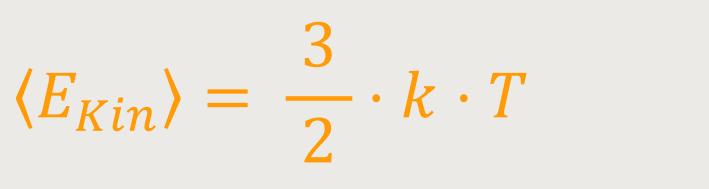
k ist der Proportionalitätsfaktor
Dieser Proportionalitätsfaktor ist die bereits angesprochene Boltzmann-Konstante. Oft wird diese in der Literatur mit kB wiedergegeben.
Die Temperatur ist demnach ein Maß für die mittlere kinetische Energie eines Moleküls in dem Gas. Egal, ob man viele Moleküle zu einem Zeitpunkt anschaut, oder einzelne Moleküle längere Zeit verfolgt. Im Mittel kommt immer diese mittlere kinetische Energie heraus. Und wenn man das jetzt einsetzt, erhält man:
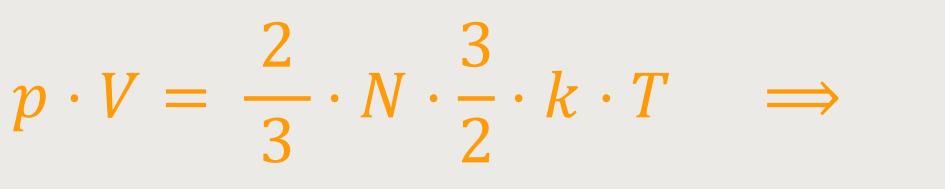

Auch hier erhalten wir die Zustandsgleichung idealer Gase.
Jetzt kann man diese Zustandsgleichung mit der Zustandsgleichung vergleichen, die wir bereits ein paar Kapitel zuvor experimentell definiert hatten:
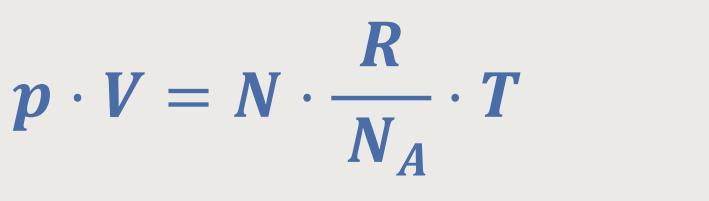
Daraus folgt jetzt, das
R ist die allgemeine Gaskonstante
NA ist die Anzahl der Moleküle in
1 kmol
Aufgrund von Untersuchungen der Brownschen Bewegung, lässt sich ermitteln, wie stark sich so ein Teilchen über längere Zeit von einem Ausgangspunkt in eine bestimmte Richtung wegbewegt. Und wenn man diese Bewegungen mittelt, lässt sich die Boltzmann-Konstante direkt feststellen.
Das R erhält man unmittelbar aus den quantitativen Untersuchungen der Zustandsgleichung idealer Gase. Und wenn man wiederum den Proportionalitätsfaktor k und die Gaskonstante R kennt, lässt sich ohne weiteres auch das NA bestimmen:

Warum ist der Proportionalitätsfaktor k so winzig? Weil es hier um die mittlere kinetische Energie nur eines einzelnen Moleküls geht. All das gilt nach wie vor für den sehr speziellen Fall eines idealen Gases, mit den entsprechenden Einschränkungen, die wir zuvor festgelegt hatten. Wir haben bisher nur drei Translations-Freiheitsgrade und damit nur die kinetische Translationsenergie zugelassen.
An dieser Stelle kann man eine erste Verallgemeinerung machen, um den Temperaturbegriff zu erweitern. Schließlich möchte man ihn ja auf verschiedene weitere Systeme anwenden. Und daher gilt, bezogen auf den zuvor gemachten Ansatz, für die mittlere kinetische Energie pro Freiheitsgrad:
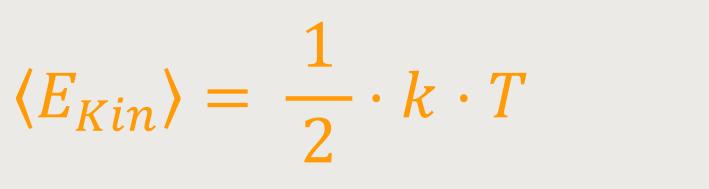
Im Allgemeinen kann man davon ausgehen, dass sich die Energien auf alle
Freiheitsgrade gleichermaßen aufteilen werden. Und damit ergibt sich eine
Verallgemeinerung. Wenn wir Moleküle betrachten, die
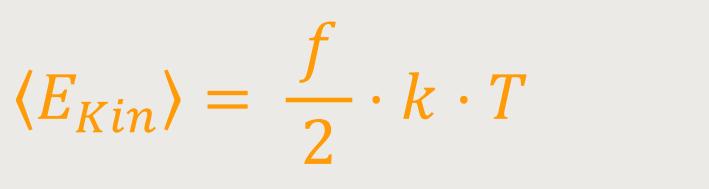
Damit sind schon deutlich allgemeinere Systeme zugelassen, bei denen eine höhere Anzahl von Freiheitsgraden vorliegen kann. Bei größeren Molekülen kommen noch Rotationsmöglichkeiten hinzu. Allerdings kann es sein, dass aufgrund quantenmechanischer Einschränkungen nicht alle Freiheitsgrade aktiviert werden können, und deshalb vielleicht nur zwei Rotations-Freiheitsgrade verbleiben.
Bei sehr hohen Temperaturen können die Moleküle zusätzlich im Innern zum Schwingen gebracht werden, wodurch ein zusätzlicher Freiheitsgrad auftritt. All das muss berücksichtigt werden. Aber ansonsten ist der Begriff der absoluten Temperatur unmittelbar verbunden mit dem Begriff der mittleren kinetischen Energie eines Moleküls des entsprechenden Körpers.
Nach wie vor muss unterschieden werden zwischen der Temperatur und der Wärmemenge. Bei der Betrachtung der Wärmemenge hatten wir bereits gesehen, dass man diese letztlich als eine Energieform auffassen kann. Es ging dabei um die gesamte kinetische Energie aller Moleküle in einem entsprechenden System. Und wenn die Masse größer ist, dann wird auch die Wärmemenge ansteigen, weil sich mehr Moleküle im System befinden.
Bei unserer Betrachtung geht es allerdings immer um die mittlere kinetische Energie eines Moleküls. Und selbst wenn man einen größeren Topf mit Wasser erhitzt, wird die mittlere kinetische Energie jedes einzelnen Moleküls gleich bleiben. Also, die Temperatur ist eine intensive Größe, die von der Masse des Körpers unabhängig ist, während die Wärmemenge die Summe aller kinetischen Energien beinhaltet. Und wenn halt mehr Moleküle vorhanden sind, dann ist auch die Summe der Energien entsprechend größer.
Jetzt machen wir noch eine zweite Verallgemeinerung, um das Thema zu erweitern und zu erläutern, was man generell unter der Temperatur versteht. Diese zweite Verallgemeinerung beinhaltet, dass wir die Definition mit f Freiheitsgraden nicht nur bei idealen Gasen anwenden, sondern auch auf Körper übertragen.
Verständlicher Weise ist das viel komplizierter zu berechnen, weil die Moleküle untereinander auch Wechselwirkungen haben. Und dann kommen noch potentielle Energien hinzu. Letztlich muss man sogar davon ausgehen, dass es zu Fluktuationen, also Schwankungen im System kommt. Denn die Moleküle werden nicht immer die gleiche kinetische Energie haben. Und so ergibt sich die Frage, wie sich die kinetische Energie oder insbesondere die Geschwindigkeiten der Moleküle in einem Gas verteilen? Also, wie viele Moleküle haben die eine Geschwindigkeit, wie viele Moleküle haben eine andere Geschwindigkeit? Und wie groß ist die anteilige Verteilung dieser Geschwindigkeiten? Diesbezüglich gibt es verschiedene Arten der Darstellung.
Nachfolgend sprechen wir kurz über die Fluktuationen und damit über die Verteilung der Geschwindigkeiten.
Der Ausgangspunkt der Betrachtung ist wieder das ideale Gas, weil es eine Sonderstellung einnimmt und besonders einfach zu beschreiben ist. Denn durch die Wechselwirkungskräfte, die in den Körpern auftreten, sei es in den Flüssigkeiten, und noch mehr bei den Festkörpern, wird die Berechnung viel aufwendiger.
Wir gehen jetzt von einem idealen Gas mit der Temperatur T aus. Des
Weiteren setzen wir hier voraus, dass sich dieses ideale Gas in einem homogenen
Gravitationsfeld befindet beispielsweise hier auf der Erdoberfläche,
mit einer Feldstärke g. Außerdem wissen wir bereits von
der barometrischen Höhenformel, dass sich der Druck in Abhängigkeit von der Höhe
bzw. der
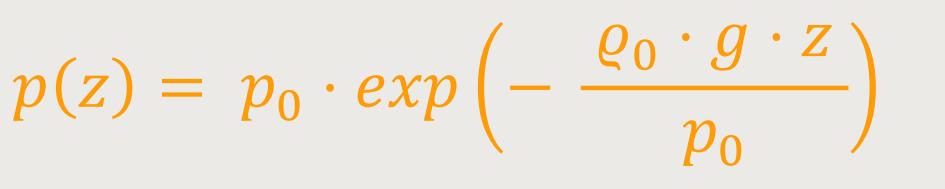
Das ist barometrische Höhenformel.
p₀ ist der Gasdruck auf einem Referenzniveau
Null
ϱ₀ ist die Dichte des Gases pro
Volumeneinheit bei Referenzniveau
g ist die Fallbeschleunigung
z ist die senkrechte Höhenkoordinate
Jetzt werden wir die barometrische Höhenformel umformen. Unter Berücksichtigung der Zustandsgleichung idealer Gase ergibt sich zunächst:
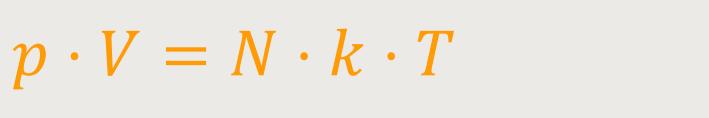
Da die Molekülanzahldichte definiert war als

Und wenn ϱ = n · m entspricht, dann erhält man nochmals durch Umformen und Einsetzen:
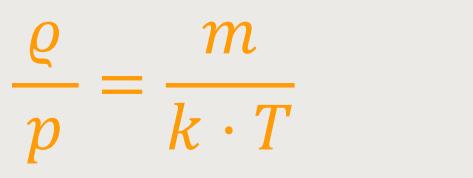
Bei einer bestimmten Temperatur ist das ϱ/p für das ideale Gas immer dasselbe, solange man die konstante Molekülmasse und die Boltzmann-Konstante hat.
Wenn man jetzt die durch Umformung erhaltene Beziehung, in die barometrische Höhenformel einsetzt, ergibt sich daraus als Folgerung:
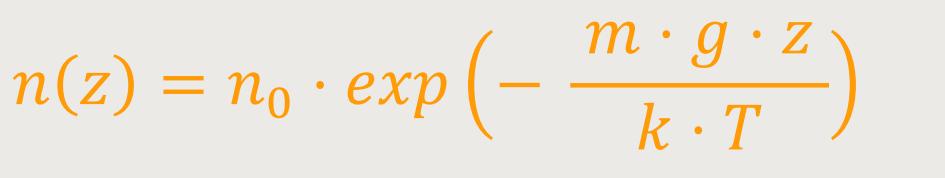
Bei aufmerksamer Betrachtung erkennt man, dass dieses
Jetzt folgt noch der Zusammenhang mit der Geschwindigkeitsverteilung. Hierzu stellen wir folgende gedankliche Überlegung an:
In einem säulenförmigen System ist die Molekülanzahldichte je nach Höhe unterschiedlich. Man kann annehmen, dass die Moleküle in der Höhe z = 0 eine derartige kinetische Energie haben, die es ihnen gerade ermöglicht, auf die Höhe m · g · z aufzusteigen, bevor sie wieder runterfallen. Und nachdem die Moleküle unterschiedlich hoch aufsteigen, gibt es entsprechend für den unteren Bereich eine Verteilung dieser Geschwindigkeitsbeträge.
Das heißt, die kinetische Energie entspricht bei der jeweiligen Temperatur:
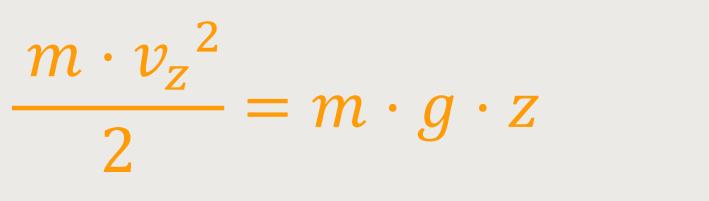
Wobei m · g · z die potentielle
Energie ist, die aufgebracht werden muss, um dieses Molekül aus der Höhe
Mittels der barometrischen Höhenformel hat man jetzt die Möglichkeit,
eine Geschwindigkeitsverteilung in der Höhe
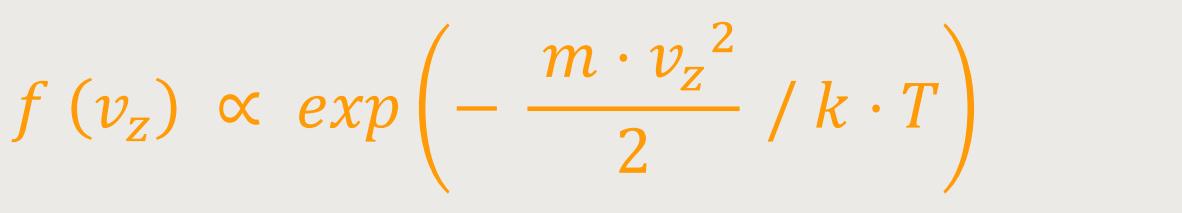
Wie lässt sich jetzt diese Verteilungsfunktion interpretieren?
Im Grunde geht es darum, dass man sich aus einer solchen Verteilungsfunktion eine
Wahrscheinlichkeit ableitet. Wie groß nämlich die
Wahrscheinlichkeit ist, dass ein Molekül eine bestimmte

Dazu muss man immer ein gewisses Intervall zulassen, in welchem die Geschwindigkeit liegen soll. Je breiter das Intervall ist, desto breiter wird auch die Wahrscheinlichkeit sein. Man sagt deshalb auch:
f (vz) ist die Wahrscheinlichkeitsdichte
Hinweis: Wenn man sich später mit der Quantenmechanik beschäftigt, wird dort die Aufenthaltswahrscheinlichkeit von Teilchen, beispielsweise von Elektronen in der Elektronenhülle eines Atoms ebenso mit einer Wahrscheinlichkeitsdichte-Funktion angegeben. Dort geht es dann um die wirklichen Wahrscheinlichkeiten, dass sich dieses Elektron in einem bestimmten Volumenelement dv befindet. Es lässt sich dann ausrechnen als Wahrscheinlichkeitsdichte multipliziert mit der Größe dieses Volumenelements dv.
Daraus ergibt sich eine wichtige Folgerung:
Wenn dieses f (vz) konkret eine
Wahrscheinlichkeitsdichte ist, dann heißt das, wenn man über
alle möglichen vz von minus unendlich bis
plus unendlich aufintegriert, muss 1 herauskommen. Denn
irgendeine Geschwindigkeit muss das Molekül ja haben. Und durch Normierung
auf 1 kann man obiger Größe noch einen entsprechenden
Vorfaktor verschaffen. Hierzu bildet man das Integral von
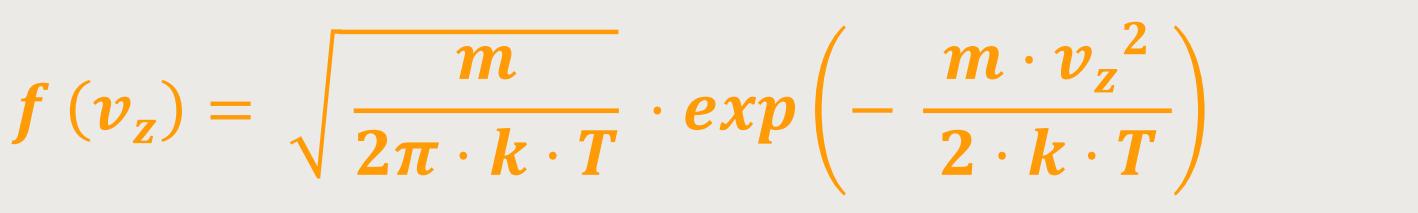
Das ist die normierte Wahrscheinlichkeitsdichte für
das Auftreten der
Bisher haben wir das nur für eine Geschwindigkeitskomponente betrachtet. In vielen Fällen ist es aber so, dass man in einem Gas ein isotropes System vorfindet. Die Moleküle haben dann in allen drei Raumrichtungen eine entsprechende Wahrscheinlichkeitsdichte. Also, wenn man daher eine solche Isotropie annimmt, kann man eine relativ einfache Verallgemeinerung machen.
Hierzu betrachtet man nicht nur ein f von einer einzelnen Komponente,
sondern von dem gesamten Geschwindigkeitsvektor, mit einer
vx-, einer vy- und einer
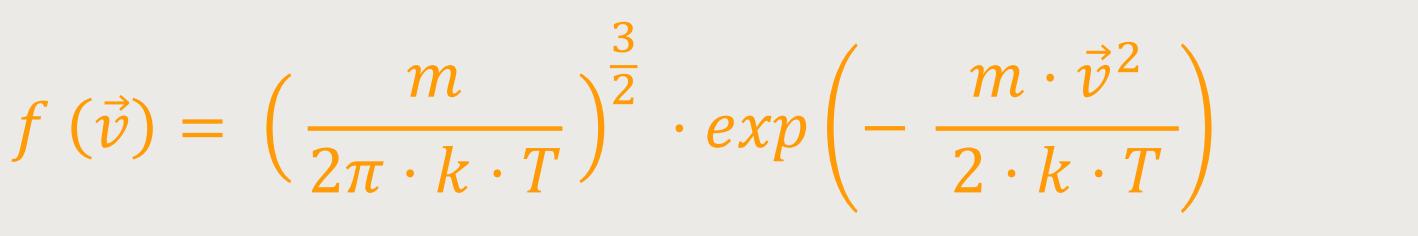
f ( )
· dvx · dvy · dvz
ist jetzt die Wahrscheinlichkeit, dass:
)
· dvx · dvy · dvz
ist jetzt die Wahrscheinlichkeit, dass:
die x-Komponente
die y-Komponente
die z-Komponente
Und dann erhält man wieder eine symmetrische Verteilung
Grafik (wird später eingefügt)
⇦ Kapitel Kapitel ⇨