
Bisher haben wir uns mit Systemen beschäftigt, in denen ideale Gase eine wesentliche Rolle gespielt haben. Jetzt sprechen über ein anderes in sich abgeschlossenes Gebiet, nämlich die realen Gase. Mittlerweile haben wir sehr oft auf die Zustandsgleichung für ideale Gase Bezug genommen:

Doch in unserer Umwelt haben wir es nur in Ausnahmefällen mit Systemen zu tun, die sich durch ideale Gase auszeichnen. In der Praxis spielen reale Gase eine größere Bedeutung.
Wenn man sich der Realität annähert, hat man eine ganz andere Situation gegenüber der, die wir bei den idealen Gasen kennengelernt hatten. Dort hatten wir ja verschiedene Idealisierungen vorausgesetzt, die in der Praxis nicht 100%ig eintreten können.
Zum einen wurde festgelegt, dass die Gasmoleküle nicht miteinander wechselwirken. Wir haben zwar ein Zusammenstoßen der Moleküle zugelassen, aber ansonsten keine Wechselwirkung zwischen den Molekülen. Zum andern haben wir vorausgesetzt, dass die Gasmoleküle ein verschwindend kleines Eigenvolumen aufweisen, also praktisch punktförmig sind. Sodass sich in dem verfügbaren Volumen nur Punkte bewegen können, ohne selbst ein Volumen einzunehmen. Diese beiden Voraussetzungen werden wir jetzt aufheben.
Wir werden jetzt einerseits davon ausgehen, dass es zwischen den Molekülen in einem solchen Gas eine gewisse Wechselwirkungskraft geben wird. Andererseits werden wir auch ein gewisses Eigenvolumen der Moleküle mit berücksichtigen. Die Moleküle sind an sich zwar nicht besonders groß, aber dennoch nehmen sie von dem ganzen Volumen im System einen entsprechenden Platz ein. Obwohl sich das im ersten Moment relativ harmlos anhört, können trotzdem deutliche Veränderungen auftreten.
Es ist natürlich nicht ganz einfach, das Eigenvolumen und die Wechselwirkungskräfte quantitativ einzuschätzen. Diese Größen hängen sehr stark von der individuellen chemischen Zusammensetzung eines Systems ab. Beim idealen Gas hat dies keine wesentliche Rolle gespielt. Egal, welches Gas man nehmen würde, ob Stickstoff, Helium oder Argon, es wäre immer die gleiche Beziehung in der Zustandsgleichung. Es bliebe auch immer dieselbe allgemeine Gaskonstante enthalten.
Wir erinnern uns, das war nichts anderes, als die Avogadro-Zahl multipliziert mit der Boltzmann-Konstante. Aber diese Größen haben nichts mit der chemischen Zusammensetzung zu tun. Nur in dem kleinen n ist in obiger Gleichung etwas enthalten, und zwar die Molzahl, hinter der aber nichts anderes steckt, als die Anzahl Kilomole der jeweiligen Substanz.
Jetzt müssen wir unseren Blickwinkel etwas ändern. Je mehr man sich der Realität annähert, desto mehr muss man das individuelle Verhalten verschiedener Substanzen mit einbeziehen. Wenn man nur die beiden vorgenannten Voraussetzungen ändert, erhalten wir bereits eine leicht veränderte Zustandsgleichung. Darüber hinaus kommt es zu einem qualitativ unterschiedlichen Verhalten der Substanzen. Sehr schnell bewegt man sich im Bereich der Phasenübergänge.
Wie zweckmäßig wäre es doch, wenn man das Molekülvolumen oder die Wechselwirkungskräfte für die verschiedenen Substanzen aus Grundgrößen herleiten könnte. Das würde vieles vereinfachen, wenn man die Werte nur in die Gleichungen einsetzten bräuchte. Aber das wird wahrscheinlich noch eine geraume Zeit dauern, bis man die Zusammenhänge besser versteht. Vielleicht wird uns ein besseres Verständnis der Quantenmechanik weiterhelfen.
Bis dahin sind wir weiterhin auf Konstanten angewiesen, die man wie in so vielen Fällen empirisch ermittelt. Das Messen der verschiedenen Substanzen in Laboratorien führt letzten Endes zu einer großen Anzahl Messdaten, die man in Tabellenbüchern und mittlerweile im Internet finden kann. Im Grunde kommt es nur darauf an zu wissen, was für Konstanten wir definieren wollen. Eine ähnliche Fragestellung hatten wir bereits in Verbindung mit der Elastizität von Körpern.
Da gab es zum Beispiel den Dehnungsmodul oder den Kompressionsmodul. In der Werkstofftechnik wünscht man sich auch, dass man sich die Moduln für die verschiedenen Substanzen ausrechnen kann. Selbst mithilfe der computergestützten physikalischen Berechnungsmethoden ist die Genauigkeit nicht besonders hoch. Für genauere Ergebnisse, greift man nach wie vor auf derartige empirische Konstanten zurück. Und so auch hier. Wir werden als erste Voraussetzung das Molekülvolumen herausgreifen.
Wenn man von einem endlichen Molekülvolumen spricht, gibt es in einem System zwangsläufig Zwischenräume, die nicht mit Molekülen angefüllt sind. Aufgrund dessen muss das zur Verfügung stehende Volumen um das tatsächliche Molekülvolumen reduziert werden, um schließlich das verbleibende Restvolumen zu ermitteln. Also das Volumen, das wir bisher in der Zustandsgleichung für ideale Gase mit V bezeichnet haben, ist jetzt ein reduziertes Volumen.
Hierfür zieht man einfach das Volumen aller Gasmoleküle in dem System ab. Man sagt auch, es verhält sich proportional zur Molzahl und zu einer bestimmten vorgegebenen Konstante für das entsprechende Gas.
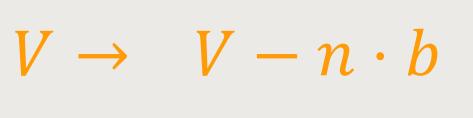
n ist die Molzahl von Kilomolen
b ist das Kovolumen (Volumen aller Moleküle pro kmol)
Das gesamte Volumen ist demnach das Volumen aller Moleküle pro kmol multipliziert mit der Anzahl der Kilomole in dem betrachteten System. Anschließend wird das Volumen der Gasmoleküle von dem Behältervolumen abgezogen. Übrig bleibt dann der freie, für die Bewegung verfügbare Raum.
Im Normalfall ziehen sich die Moleküle an. Wäre das nicht der Fall, würde vor allem im Falle einer dichteren Anordnung der Moleküle beispielsweise in einer Flüssigkeit, das Medium nicht zusammenhalten. Bei einem Festkörper muss es natürlich ebenfalls Wechselwirkungen anziehender Art geben, die dem Körper eine feste Form verleihen. Diese Anziehungskraft zwischen den Molekülen ist auch in geringem Umfang bei Gasen vorhanden.
Die Wechselwirkungskräfte haben einen Einfluss auf den Druck in einem System. Der Druck kommt dadurch zustande, dass die Moleküle wie ein Trommelfeuer auf die Gefäßwände prallen. Solange man von einem idealen Gas spricht, hält sich das Alles im Rahmen. Wenn man hingegen ein reales Gas betrachtet, dann liegt eine ähnliche Situation vor, wie bei der Oberflächenspannung in einer Flüssigkeit.
Dadurch, dass die Moleküle miteinander in Wechselwirkung stehen, werden Moleküle, die sich in Wandnähe befinden, in irgendeiner Weise von den dahinterliegenden Nachbarmolekülen durch deren Anziehungskraft zurückgehalten. Sie prallen mit einem etwas geringeren Impuls auf die Wand. Dadurch wird der Impulsübertrag kleiner, und das bedeutet einen geringeren Druck. Also muss man sich überlegen, wie groß diese Reduktion des messbaren Druckes an einer Gefäßwand oder an der Membrane eines Druckgebers sein wird.
Um der Lösung ein Stück näher zu kommen, kann man zunächst die Anziehungskraft nur eines Moleküls betrachten. Also ein Molekül, das sich bereits in Wandnähe befindet, und unmittelbar dagegen prallen wird. Dadurch wird ein Impulsübertrag bewirkt, der dann letztlich den Druck hervorruft, der auf diese Wand ausgeübt wird.
Diese Anziehungskraft verhindert, dass ein wandnahes Molekül mit voller Wucht gegen die Wand prallt. Jetzt hat man aber nicht nur ein einzelnes Molekül, das für den Druck verantwortlich ist, sondern es wird eine große Anzahl von Molekülen pro Zeiteinheit auf die Behälterwand treffen. Diese Anzahl ist natürlich auch wieder proportional zu der Molekülanzahldichte in dem Gas. Je größer die Anzahl pro Volumeneinheit, desto mehr treffen auf die Wand.
Dadurch ergibt sich, dass der Druck, der gemessen wird, kleiner ist als der Druck, der eigentlich im Innern des Systems vorhanden ist. Denn gemessen wird immer nur an einer Wand bzw. einer Membrane. Aber im Inneren des Gases, wo sich die Gasmoleküle frei bewegen können, und von anderen Molekülen umgeben sind, fällt die einseitige Kraftwirkung weg. Wenn der Druck im Innern also größer wird, ergibt sich für den Druck
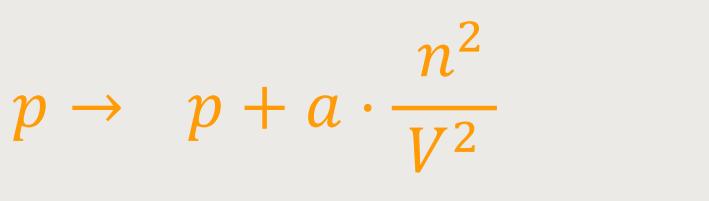
a ist ein Proportionalitätsfaktor
a · n²/V² nennt man den Binnendruck
Also, das Volumen, das wirklich für die Bewegung der Moleküle zur Verfügung steht, ist kleiner als das Volumen des Gefäßes. Und der Druck, den die Moleküle im Inneren ausüben, ist größer als der Druck, den man durch irgendeinen Sensor misst.
Mit diesen beiden Erweiterungen für p und V lässt sich jetzt die eingangs erwähnte Zustandsgleichung in eine praxisnahe Form bringen. Als Basis dient weiterhin die vereinfachte Zustandsgleichung für ideale Gase, aber jetzt ergänzen wir entsprechend, indem wir einsetzen:
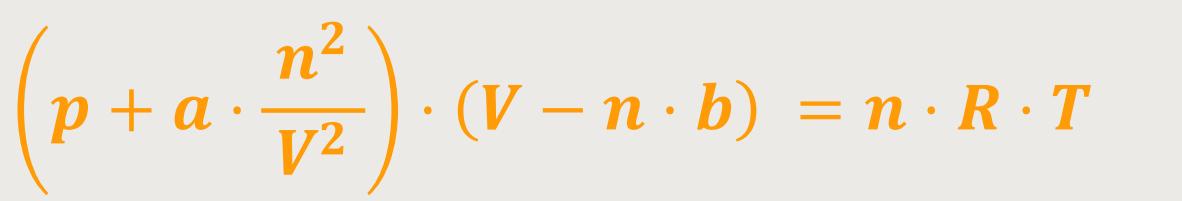
Das ist die Van-der-Waalssche Zustandsgleichung.
Diese Gleichung sieht ohne Frage anders aus, als die bisher verwendete, und sie lässt
auch auf einen anderen Zusammenhang zwischen
Mittels eines
Bei den Van-der-Waals-Gasen wird
Wenn man sich im Bereich von hohen Temperaturen
Laut
Trotzdem können ab einem bestimmten Bereich Merkwürdigkeiten
auftreten, die nicht hyperbelähnlich verlaufen. Bei der Untersuchung
realer Systeme liegt das daran, dass in dem abweichenden Bereich offensichtlich ein
Phasenübergang stattfindet. In einem
Je geringer aber die Temperaturen werden, desto mehr verändert sich der Verlauf der Isothermen. Jede weitere Isotherme, die diesen Bereich schneidet, verläuft dann waagerecht weiter und wenn sie aus dem Bereich wieder austritt, setzt sie den hyperbelähnlichen Verlauf fort. Wie lässt sich das interpretieren?
Das System wechselt von einem homogenen in ein inhomogenes System über. In dem Bereich, wo die Kurven quasi eine nicht physikalische Welle zeigen, spricht man von einem Bereich der Zweiphasigkeit. Je nach Temperatur, wechselt das System bei einem gewissen Druck, von dem einen in das andere System. Oberhalb hingegen bleibt es bei dem homogenen einphasigen Bereich. Der entscheidende Punkt ist hierbei, dass während dieser Zweiphasigkeit eine gasförmige Phase und eine flüssige Phase nebeneinander koexistieren. Was heißt das im Detail?
Nehmen wir an, wir hätten ein großes Volumen und entsprechend einen kleinen Druck. Jetzt verringern wir das Volumen des Gases, indem wir zum Beispiel Wasserdampf komprimieren. Daraufhin wird der tatsächliche Druck dieses Dampfes in dem System ansteigen. Irgendwann kommt der Punkt, wo das nicht mehr ohne weiteres fortgesetzt werden kann. Die Moleküle lassen sich nicht mehr weiter komprimieren. Demzufolge beginnt jetzt ein Phasenübergang von dem gasförmigen Wasserdampf zum flüssigen Wasser.
Man kann sagen, sobald es irgendwelche Störstellen gibt, beispielsweise Ionen, beginnt die Bildung der neuen Phase. Meistens geschieht das mithilfe von irgendwelchen kleinen Molekül-Clustern, die sich plötzlich zusammenfügen, und die dann Ausgangspunkt für die Formierung der neuen Phase sind. Diese Keimbildung oder „Nukleation” vollzieht sich sehr schnell, und ist der erste Teilprozess, der einen Phasenübergang erster Ordnung einleitet.
Doch wenn man das System weiter komprimiert, wird der Druck nicht mehr ansteigen. Vielmehr bleibt der Druck eine Zeit lang konstant. Man spricht hier auch von einem sogenannten Sättigungsdruck. Je kleiner die Temperatur ist, desto kleiner ist auch der Sättigungsdruck. Während das System diese Gleichgewichtszustände durchläuft, wird von dem Gas immer mehr in Flüssigkeit übergehen, und der Dampf wird weiter abnehmen.
Während dieses Prozesses teilt sich der durchschrittene Bereich in zwei Teilabschnitte auf. Ein Teil ist bereits verflüssigt, wogegen der andere Teil noch dampfförmig ist. Ab einem bestimmten Punkt ist der gesamte Dampf in Flüssigkeit übergegangen. Es gilt zu berücksichtigen, dass sich weder die Anzahl der Moleküle, noch die Temperatur geändert haben. Und auch der Druck ist konstant geblieben, wenngleich es sich hierbei um den Sättigungsdruck der Flüssigkeit in dem System handelt.
Hierzu kann man sich einen senkrecht stehenden Zylinder mit einem Kolben vorstellen. Die untere Hälfte des Volumens enthält Flüssigkeit, wogegen die obere Hälfte mit Wasserdampf gefüllt ist. Das System hat zudem eine bestimmte Temperatur. Wenn man das Volumen des Dampfes ändert, wird sich der Druck nicht ändern. Das System kann nicht mehr als diesen Sättigungsdampfdruck herstellen. Selbst wenn man mit einem Kolben den Dampf weiter komprimiert, werden etliche Moleküle in den flüssigen Zustand hineinkondensieren. Aber der Druck wird dadurch nicht steigen.
Man könnte hier von einer maximalen Spannkraft sprechen, die diese Flüssigkeit gegen den Dampf hat. Es herrscht quasi bei einer bestimmten Temperatur ein Gleichgewichtszustand zwischen Flüssigkeit und Dampf. Das Ganze funktioniert solange, bis nur noch Flüssigkeit vorhanden ist, und man schließlich mit dem Kolben an der Flüssigkeitsoberfläche anstößt. An dieser Stelle lässt sich das System nur noch mit sehr hohem Aufwand komprimieren, weshalb der Druck auch steil ansteigen wird. Je niedriger die Temperatur ist, desto steiler steigt der Druck an.
Im Bereich des Zweiphasengebietes gibt es eine Koexistenz von flüssiger und
gasförmiger Phase. Diese zwei Koexistenzen werden im Van-der-Waals-Diagramm
durch eine Wellenlinie dargestellt. Vom Anfangspunkt bis zum Endpunkt
verläuft eine waagerechte Schnittlinie. Diese Grenzlinie entspricht
dem bestimmten Sättigungsdruck bei einer bestimmten Temperatur. Durch die
Wellenlinie entsteht einerseits ein Wellental mit einer Teil
Die Van-der-Waals-Gleichung liefert nur die Wölbung bestehend aus dem Minimum und
Maximum. Wenn man die tatsächliche Gleichgewichtsdampflinie sucht, so
ist das die Waagerechte, bei der die beiden Teilflächen gleich groß sind.
Jetzt hat diese Darstellung aber auch noch eine dritte Dimension, nämlich die
Temperatur. Und so lässt das auch
Hierzu nimmt man wieder das
Wenn man die Temperatur als dritte Achse in ein
Es bietet sich auch die Möglichkeit, das
Aber dann gibt es noch eine zweite stark ansteigende Fläche, die sich bildet, wenn der Übergang zwischen flüssig und fest stattfindet. Diese Linie, von der alle Flächen weglaufen nennt man „Tripellinie”, weil hier die Zustände gasförmig, flüssig und fest in Koexistenz sind. Und wenn man sich die Linienverläufe anschaut, stoßen alle am Tripelpunkt zusammen.
Die Linie vom Tripel
Bei niedrigen Temperaturen und hohen Drücken ist das System fest. Bei höheren Temperaturen wird das System flüssig. Und wenn die Drücke kleiner werden, bildet sich im System Dampf. So laufen die Phasen im Wesentlichen ab.
Wasser (H₂O) zeigt in einem solchen Phasendiagramm eine gewisse Anomalität. Wenn man die Koexistenz-Linie zwischen flüssig und fest betrachtet, kann man erkennen, dass bei zunehmendem Druck die Temperatur sogar zurückgeht. Wenn man zum Beispiel Eis einem starken Druck aussetzt, dann schmilzt es, was bei Kohlendioxid nicht der Fall ist. Wasser hat somit eine Anomalie, die für uns lebenswichtig ist.
Bei
Je nach Material gibt es bei einem bestimmten Druck mit einer bestimmten Temperatur einen kritischen Punkt. Unterhalb dieses Punktes hat man bei konstanten niedrigeren Temperaturen Phasenübergänge. Aber über dem kritischen Punkt gibt es keine flüssige Phase mehr. Es gibt allerdings eine Möglichkeit, einen Phasenübergang zu umgehen. Dazu muss man die Temperatur jeweils anpassen und oberhalb der kritischen Temperatur halten. Man umgeht damit den Zweiphasenbereich und hat trotzdem die ganze Zeit eine flüssige Phase und bleibt ständig in einem einphasigen Gebiet.
Alternativ könnte man auch bei konstantem Volumen die Temperatur und damit den Druck erhöhen. Wenn man das an der Stelle des kritischen Punkt durchläuft, hat man die ganze Zeit über einen Zweiphasenbereich. Unten liegt die Flüssigkeit und oberhalb befindet sich das Gas, bis man schließlich den kritischen Punkt durchstößt.
⇦ Kapitel Kapitel ⇨